
Wat zijn stofwisselingsziekten?
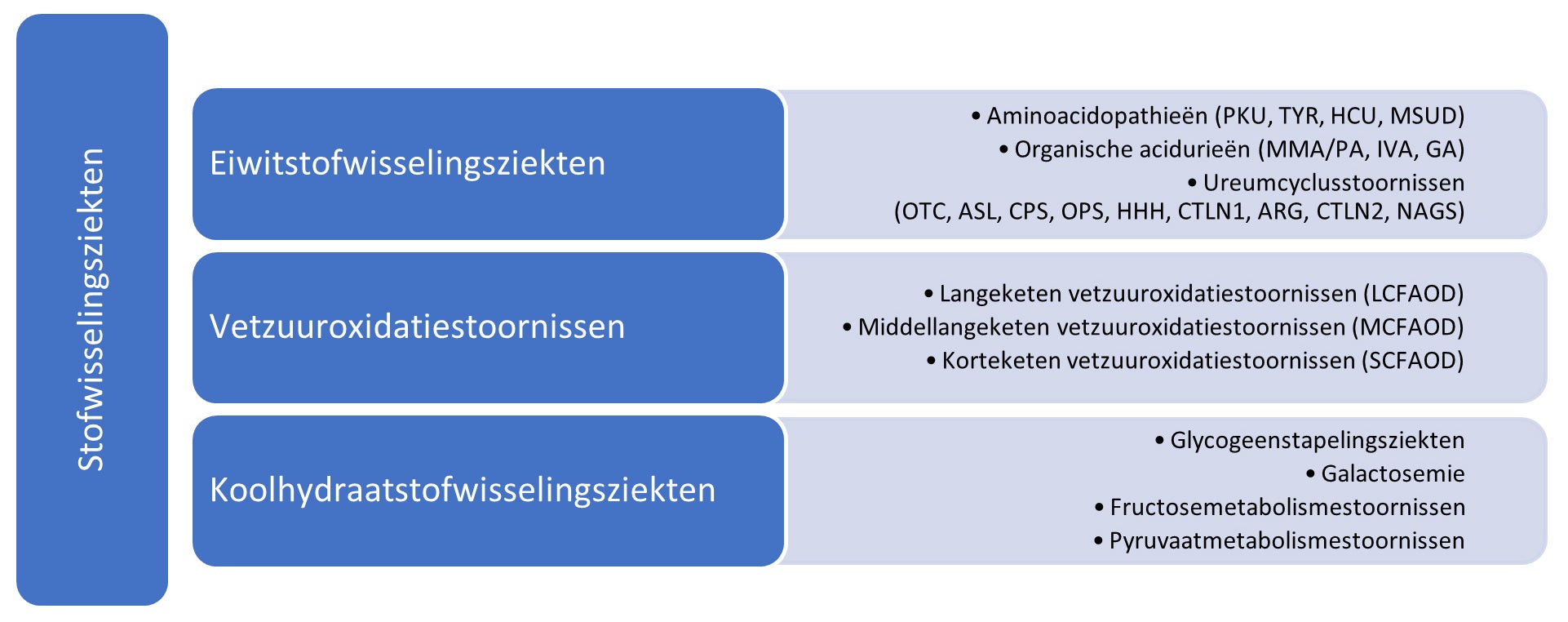
Stofwisselingsziekten zijn aangeboren genetische aandoeningen waarbij een specifiek enzymdefect het normale eiwit-, vet- of koolhydraatmetabolisme verstoort. Door een verminderde of afwezige enzymactiviteit in deze aandoeningen, stapelen specifieke stoffen zich op in het lichaam in toxische hoeveelheden. Dit kan er ook tot leiden dat het lichaam niet in staat is om voldoende stoffen dat het nodig heeft aan te maken, wat kan leiden tot tekorten. Indien deze metabole stoornissen onbehandeld blijven, kunnen zij resulteren in een reeks medische- en ontwikkelingsstoornissen, van cognitieve beperkingen tot orgaanfalen en zelfs de dood.
Veel negatieve gevolgen van stofwisselingsziekten kunnen beperkt worden door een vroege opsporing en begeleiding welke kan bestaan uit medicinale en/of dieetinterventie met behulp van voeding voor medisch gebruik.
Individueel zijn stofwisselingsziekten zeldzaam maar samen komen ze vaker voor (3,5 – 5,9% van de wereldwijde populatie (1)). Vele stofwisselingsziekten worden opgespoord bij de geboorte met behulp van de hielpriktest, een screening bij pasgeborenen. Dat maakt een vroege identificatie en initiatie van de begeleiding mogelijk wat de uitkomsten van patiënten kan verbeteren. De effectiviteit van screening bij pasgeborenen heeft geleid tot een stijging van de incidentiecijfers van stofwisselingsziekten en hoewel screening aanbevolen is, wordt dit niet uitgevoerd in alle landen.
Fenylketonurie (PKU) is één van de meest voorkomende stofwisselingsziekten die een aangepaste dieetbegeleiding vereisen. De aandoening heft een globale incidentieratio van ongeveer 1 op 10.000 tot 1 op 12.000 (2). PKU is een aangeboren aandoening waarbij het PKU gen geërfd wordt van beide ouders. Het is niet gelinkt aan geslacht, dit noemt men autosomal recessief. PKU is een eiwitstofwisselingsziekte, specifiek gerelateerd aan het aminozuur fenylalanine (aminozuren zijn de bouwstenen van eiwitten). PKU kenmerkt zich aan de hand van een deficiëntie van het hepatisch enzym fenylalanine hydroxylase dat fenylalanine (Phe) omzet in tyrosine (Tyr), wat resulteert in een opstapeling van Phe en een tekort aan Tyr in het bloed. Indien onbehandeld kan dit leiden tot toxische Phe-waarden wat onherstelbare hersenschade kan veroorzaken. Dit kan leerproblemen, gedragsveranderingen, trillingen en epileptische aanvallen teweegbrengen. Lage Tyr-waarden veroorzaken een verstoorde neurotransmitterproductie en een melaninetekort, wat zich uit als een karakteristieke bleke teint. Zowel Phe als Tyr waarden moeten gemonitord worden bij PKU door regelmatig bloedstalen te nemen en door de inname van natuurlijk eiwit te beperken. Omdat Phe een essentieel aminozuur is, is het noodzakelijk voor het normaal functioneren van het lichaam. Het lichaam kan dit zelf niet aanmaken. Om te voorkomen dat Phe zich opstapelt in toxische hoeveelheden, moet een kleine, goed afgewogen hoeveelheid natuurlijk eiwit geconsumeerd worden zodat aan de Phe behoeften voldaan maar niet overschreden wordt. Personen met PKU hebben verschillende Phe-toleranties afhankelijk van de ernst van hun aandoening, leeftijd en groei. Eiwitten zijn essentieel voor de groei en herstel van het lichaam. Kleine hoeveelheden natuurlijk eiwit zijn onvoldoende voor PKU-patiënten om te voldoen aan hun totale eiwitbehoeftes. Producten die aminozuurpreparaten genoemd worden, worden voorgeschreven zodat patiënten aan hun vereiste eiwitbehoeften kunnen voldoen. Verschillende termen worden gebruikt om aminozuurpreparaten te omschrijven zoals aminozuurmengsels, eiwitvervangers, medische voeding of dieetvoeding. Aminozuurpreparaten bevatten ‘eiwitequivalent’, wat inhoudt dat het product alle aminozuren in eiwitten bevat, behalve of in slechts zeer beperkte hoeveelheid het schadelijke aminozuur, in dit geval Phe. Bijkomende nutriënten zijn toegevoegd aan aminozuurpreparaten om nutritionele tekorten te voorkomen omwille van het restrictieve karakter van hun dieet. Er bestaan 2 voorname soorten van aminozuurpreparaten voor PKU – aminozuurmengsels op basis van aminozuren en GMP-aminozuurpreparaten op basis van glycomacropeptide (GMP). Eiwitarme voedingsmiddelen die geproduceerd worden als alternatief voor normale voedingsmiddelen zijn beschikbaar voor de dieetbegeleiding van PKU, of andere eiwitstofwisselingsziekten. Dit zijn basisvoedingsmiddelen zoals eiwitarm brood, eiwitarme pasta en melkvervangers die niet enkel normaliteit maar ook extra energie aan het dieet kunnen toevoegen om de algehele metabole controle te ondersteunen. Een vroege diagnose en dieetbegeleiding kunnen patiëntenuitkomsten significant verbeteren (3). Meer informatie over stofwisselingsziekten en hun dieetbegeleiding kan gevonden worden op Vitaflo | Nestlé Health Science (nestlehealthscience.com).
Iedereen met een stofwisselingsziekte die meer informatie wenst over de aandoening, de begeleiding en de verwachtingen, zou medisch advies moeten zoeken van zijn of haar arts en/of diëtist.
Fenylketonurie (PKU) is één van de meest voorkomende stofwisselingsziekten die een aangepaste dieetbegeleiding vereisen. De aandoening heft een globale incidentieratio van ongeveer 1 op 10.000 tot 1 op 12.000 (2). PKU is een aangeboren aandoening waarbij het PKU gen geërfd wordt van beide ouders. Het is niet gelinkt aan geslacht, dit noemt men autosomal recessief. PKU is een eiwitstofwisselingsziekte, specifiek gerelateerd aan het aminozuur fenylalanine (aminozuren zijn de bouwstenen van eiwitten). PKU kenmerkt zich aan de hand van een deficiëntie van het hepatisch enzym fenylalanine hydroxylase dat fenylalanine (Phe) omzet in tyrosine (Tyr), wat resulteert in een opstapeling van Phe en een tekort aan Tyr in het bloed. Indien onbehandeld kan dit leiden tot toxische Phe-waarden wat onherstelbare hersenschade kan veroorzaken. Dit kan leerproblemen, gedragsveranderingen, trillingen en epileptische aanvallen teweegbrengen. Lage Tyr-waarden veroorzaken een verstoorde neurotransmitterproductie en een melaninetekort, wat zich uit als een karakteristieke bleke teint. Zowel Phe als Tyr waarden moeten gemonitord worden bij PKU door regelmatig bloedstalen te nemen en door de inname van natuurlijk eiwit te beperken. Omdat Phe een essentieel aminozuur is, is het noodzakelijk voor het normaal functioneren van het lichaam. Het lichaam kan dit zelf niet aanmaken. Om te voorkomen dat Phe zich opstapelt in toxische hoeveelheden, moet een kleine, goed afgewogen hoeveelheid natuurlijk eiwit geconsumeerd worden zodat aan de Phe behoeften voldaan maar niet overschreden wordt. Personen met PKU hebben verschillende Phe-toleranties afhankelijk van de ernst van hun aandoening, leeftijd en groei. Eiwitten zijn essentieel voor de groei en herstel van het lichaam. Kleine hoeveelheden natuurlijk eiwit zijn onvoldoende voor PKU-patiënten om te voldoen aan hun totale eiwitbehoeftes. Producten die aminozuurpreparaten genoemd worden, worden voorgeschreven zodat patiënten aan hun vereiste eiwitbehoeften kunnen voldoen. Verschillende termen worden gebruikt om aminozuurpreparaten te omschrijven zoals aminozuurmengsels, eiwitvervangers, medische voeding of dieetvoeding. Aminozuurpreparaten bevatten ‘eiwitequivalent’, wat inhoudt dat het product alle aminozuren in eiwitten bevat, behalve of in slechts zeer beperkte hoeveelheid het schadelijke aminozuur, in dit geval Phe. Bijkomende nutriënten zijn toegevoegd aan aminozuurpreparaten om nutritionele tekorten te voorkomen omwille van het restrictieve karakter van hun dieet. Er bestaan 2 voorname soorten van aminozuurpreparaten voor PKU – aminozuurmengsels op basis van aminozuren en GMP-aminozuurpreparaten op basis van glycomacropeptide (GMP). Eiwitarme voedingsmiddelen die geproduceerd worden als alternatief voor normale voedingsmiddelen zijn beschikbaar voor de dieetbegeleiding van PKU, of andere eiwitstofwisselingsziekten. Dit zijn basisvoedingsmiddelen zoals eiwitarm brood, eiwitarme pasta en melkvervangers die niet enkel normaliteit maar ook extra energie aan het dieet kunnen toevoegen om de algehele metabole controle te ondersteunen. Een vroege diagnose en dieetbegeleiding kunnen patiëntenuitkomsten significant verbeteren (3). Meer informatie over stofwisselingsziekten en hun dieetbegeleiding kan gevonden worden op Vitaflo | Nestlé Health Science (nestlehealthscience.com).
Iedereen met een stofwisselingsziekte die meer informatie wenst over de aandoening, de begeleiding en de verwachtingen, zou medisch advies moeten zoeken van zijn of haar arts en/of diëtist.
Referenties:
1. Nguengang Wakap S, Lambert DM, Olry A, Rodwell C, Gueydan C, Lanneau V, et al. Estimating cumulative point prevalence of rare diseases: analysis of the Orphanet database. European Journal of Human Genetics 2019 28:2 [Internet]. 2019 Sep 16 [cited 2021 Oct 21];28(2):165–73. Available from: https://www.nature.com/articles/s41431-019-0508-0
2. Dixon M, MacDonald A, White F, Stafford J. Disorders of Amino Acid Metabolism, Organic Acidaemias and Urea Cycle Disorders. Clinical Paediatric Dietetics: Fourth Edition [Internet]. 2014 Nov 17 [cited 2021 Oct 13];381–525. Available from: https://onlinelibrary.wiley.com/doi/full/10.1002/9781118915349.ch17
3. Channon S, Goodman G, Zlotowitz S, Mockler C, Lee PJ. Effects of dietary management of phenylketonuria on long-term cognitive outcome. Archives of Disease in Childhood [Internet]. 2007 Mar 1 [cited 2021 Oct 15];92(3):213–8. Available from: https://adc.bmj.com/content/92/3/213